
Creating phylogenetic trees from dna sequences answers – Creating phylogenetic trees from DNA sequences provides a powerful approach to unraveling the intricate tapestry of evolutionary relationships among organisms. This technique harnesses the wealth of genetic information encoded within DNA to reconstruct the branching patterns that connect species over time, enabling scientists to explore the origins and diversification of life on Earth.
Phylogenetic trees, rooted in the principles of molecular biology and computational analysis, serve as invaluable tools for understanding the genetic relatedness of species, tracing the evolution of traits, and investigating the historical events that have shaped the diversity of life.
Phylogenetic Tree Construction

Phylogenetic trees are diagrams that represent the evolutionary relationships among different species or other taxa. They are used to infer the history of life on Earth and to understand the processes that have shaped the diversity of life.
There are a number of different methods used to construct phylogenetic trees from DNA sequences. The most common method is called maximum parsimony, which seeks to find the tree that requires the fewest evolutionary changes. Other methods include neighbor-joining, which uses a distance-based approach, and Bayesian inference, which uses a probabilistic approach.
Software and Online Tools for Phylogenetic Tree Construction
- MEGA
- PAUP*
- RAxML
- PhyML
- MrBayes
DNA Sequence Alignment
DNA sequence alignment is the process of aligning two or more DNA sequences to identify regions of similarity and difference. This is an important step in phylogenetic tree construction, as it allows researchers to compare the sequences of different species and identify the changes that have occurred over time.
There are a number of different methods used for DNA sequence alignment. The most common method is called pairwise alignment, which aligns two sequences at a time. Other methods include multiple sequence alignment, which aligns three or more sequences at a time, and progressive alignment, which aligns sequences in a step-by-step process.
Software and Online Tools for DNA Sequence Alignment
- ClustalW
- T-Coffee
- Muscle
- MAFFT
- EMBOSS
Model Selection and Parameter Estimation

Model selection and parameter estimation are important steps in phylogenetic tree construction. Model selection involves choosing the best model of evolution to use for the data. Parameter estimation involves estimating the values of the parameters in the model.
There are a number of different models of evolution that can be used for phylogenetic tree construction. The most common model is the Jukes-Cantor model, which assumes that all nucleotide substitutions are equally likely. Other models include the Kimura 2-parameter model, which takes into account the different rates of substitution for different types of nucleotides, and the Hasegawa-Kishino-Yano model, which takes into account the different rates of substitution for different codon positions.
Parameter estimation involves estimating the values of the parameters in the model. These parameters include the substitution rate, the transition/transversion ratio, and the proportion of invariable sites.
How to Use Software to Perform Model Selection and Parameter Estimation
There are a number of different software programs that can be used to perform model selection and parameter estimation. Some of the most common programs include MEGA, PAUP*, and MrBayes.
To perform model selection using MEGA, follow these steps:
- Open the MEGA program.
- Click on the “Phylogenetics” menu.
- Select the “Model Selection” option.
- Choose the model of evolution that you want to use.
- Click on the “OK” button.
To perform parameter estimation using MEGA, follow these steps:
- Open the MEGA program.
- Click on the “Phylogenetics” menu.
- Select the “Parameter Estimation” option.
- Choose the model of evolution that you want to use.
- Click on the “OK” button.
Tree Evaluation and Interpretation: Creating Phylogenetic Trees From Dna Sequences Answers
Tree evaluation and interpretation are important steps in phylogenetic tree construction. Tree evaluation involves assessing the accuracy of the tree. Tree interpretation involves using the tree to make inferences about the evolutionary history of the taxa.
There are a number of different methods used to evaluate phylogenetic trees. The most common method is called bootstrapping, which involves resampling the data and reconstructing the tree multiple times. Other methods include jackknifing and Bayesian posterior probabilities.
Tree interpretation involves using the tree to make inferences about the evolutionary history of the taxa. This can be done by looking at the branching patterns of the tree, the lengths of the branches, and the positions of the taxa on the tree.
How to Use Software to Evaluate and Interpret Phylogenetic Trees, Creating phylogenetic trees from dna sequences answers
There are a number of different software programs that can be used to evaluate and interpret phylogenetic trees. Some of the most common programs include MEGA, PAUP*, and MrBayes.
To evaluate a phylogenetic tree using MEGA, follow these steps:
- Open the MEGA program.
- Click on the “Phylogenetics” menu.
- Select the “Tree Evaluation” option.
- Choose the method of evaluation that you want to use.
- Click on the “OK” button.
To interpret a phylogenetic tree using MEGA, follow these steps:
- Open the MEGA program.
- Click on the “Phylogenetics” menu.
- Select the “Tree Interpretation” option.
- Choose the method of interpretation that you want to use.
- Click on the “OK” button.
Applications of Phylogenetic Trees
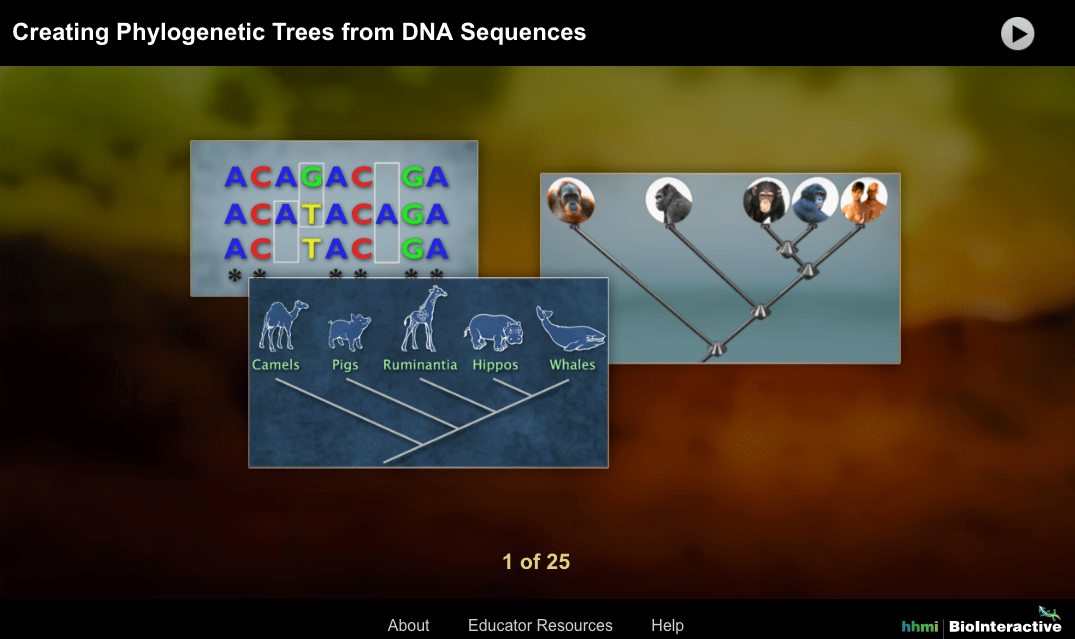
Phylogenetic trees have a wide range of applications in various fields. They are used to study the evolution of species, to identify the relationships between different groups of organisms, and to understand the processes that have shaped the diversity of life on Earth.
Some of the specific applications of phylogenetic trees include:
- Taxonomy:Phylogenetic trees are used to classify organisms into different groups, such as species, genera, and families.
- Evolutionary biology:Phylogenetic trees are used to study the evolution of species and to understand the processes that have shaped the diversity of life on Earth.
- Medicine:Phylogenetic trees are used to study the evolution of diseases and to develop new treatments.
- Agriculture:Phylogenetic trees are used to study the evolution of crops and to develop new varieties.
- Conservation biology:Phylogenetic trees are used to identify endangered species and to develop conservation strategies.
Case Studies
There are many examples of how phylogenetic trees have been used to answer scientific questions. Here are a few case studies:
- The origin of life:Phylogenetic trees have been used to study the origin of life on Earth. By comparing the DNA sequences of different organisms, scientists have been able to infer the relationships between different groups of organisms and to trace their evolutionary history back to a common ancestor.
- The evolution of humans:Phylogenetic trees have been used to study the evolution of humans. By comparing the DNA sequences of humans and other primates, scientists have been able to infer the relationships between different groups of humans and to trace their evolutionary history back to a common ancestor.
- The spread of diseases:Phylogenetic trees have been used to study the spread of diseases. By comparing the DNA sequences of different strains of a virus, scientists have been able to track the spread of the virus and to identify the source of the outbreak.
General Inquiries
What is a phylogenetic tree?
A phylogenetic tree is a diagram that represents the evolutionary relationships among different species or other taxa. It is based on the principle that species that share more recent common ancestors are more closely related to each other than species that share more distant common ancestors.
How are phylogenetic trees created?
Phylogenetic trees are created by comparing the DNA sequences of different species. The more similar the DNA sequences, the more closely related the species are likely to be. Various methods, such as maximum parsimony, neighbor-joining, and Bayesian inference, are used to analyze the data and construct the tree.
What are the applications of phylogenetic trees?
Phylogenetic trees have a wide range of applications in biology, including taxonomy, systematics, evolutionary biology, conservation biology, and medicine. They can be used to study the evolution of traits, identify common ancestors, and infer the historical events that have shaped the diversity of life.
